

Peter Nowell and David Hungerford in 1960
Chronic Myeloid Leukemia (CML) is not a cancer you hear about very often. That’s not because it isn’t serious — until recently, it carried a frightening prognoses. Until about 2000, newly diagnosed CML patients had a 5-year survival rate of 31%. But we don’t hear much about CML because it is a rare disease, and affects far fewer people than cancers of the breast or prostate, or lung cancer. The American Cancer Society estimated there were almost 1.7 million new cancer cases in 2017, of which CML comprised only about 8,400 (one in 200). But survival statistics for patients with CML have undergone a profound change in the past 15 years, as patients have been treated with a revolutionary new drug called Gleevec (Glivec in Europe). Almost all of these patients are still alive.
It started with an observation
The story of the new treatment for CML began, as so many biomedical stories do, with a surprising observation. This was reported in 1960 by two scientists from the Fox Chase Cancer Research Institute in Philadelphia, Peter Nowell and his young colleague David Hungerford. Using a relatively new technique that had enabled the reliable visualization of human chromosomes, they had observed a singular correlation: in almost every case, the leukemic cells of CML patients had a malformation of one of their chromosomes. One of the two chromosomes 22 in their cancer cells was shorter than normal.
In 1960, human cytogenetics, the microscopic analysis of chromosomes, was in its infancy. Watson and Crick’s double helix model for the structure of DNA was only 7 years old. The correct number of human chromosomes (23 pairs) had been determined just 4 years earlier. But the chromosomes could now be reliably identified by their shape and size. They were numbered in order of decreasing size, with number 1 being the largest, and number 22 a runt containing only 1.5% of human DNA. At the time, only the male-specific chromosome Y was thought to be smaller than 22. Later, it turned out that chromosome number 21 was actually a little smaller than 22, but the numbering was never changed, to avoid confusion.
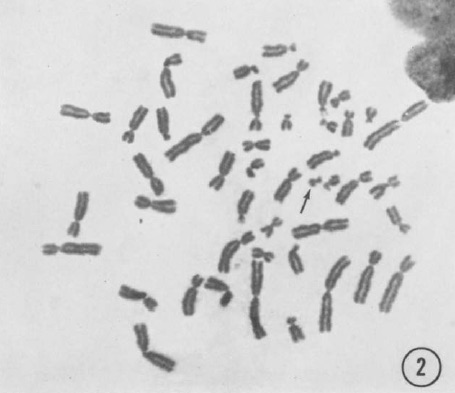
What made Nowell and Hungerford’s observation even more striking was that chromosome 22 in the healthy cells of the CML patients were normal-looking; only one of the two chromosomes 22 of their leukemic cells was altered. This observation was quickly confirmed, and to this day shortened chromosome 22, known as the “Philadelphia chromosome”, is a key diagnostic feature of CML.
Fishing expeditions
In the pictures of chromosomes available in 1960 the material of all the chromosomes was undifferentiated. Only their different lengths distinguished them (Figure 1). The difference in chromosome 22 of CML cells remained an unexplained, if diagnostically valuable, observation. As is so often the case, figuring out what had happened depended on a new technique, and a contributor with a new approach.
Dr. Janet Davison Rowley was that person. A precocious teenager, Janet Davison obtained a bachelor’s degree from the University of Chicago at age 19. She then became the only woman in her medical school class, graduating in 1948. She was 23. The next day she married Dr. Donald Adams Rowley. Both completed their internships in 1951. She spent the next twenty years raising four boys while working three days a week at various jobs, including a clinic for children with Down Syndrome at the University of Chicago. In that work, she was well aware of the importance of chromosomal abnormalities; for example, children with Down syndrome have 3 copies of chromosome 21.
In 1962, the Rowleys were in England on sabbatical. Janet Rowley spent the year at Oxford University, where she learned techniques of chromosomal microscopy, or cytogenetics. Back in Chicago, she was offered a position that included some laboratory space, a microscope, a darkroom for developing film, and a salary of $5,000 a year. A breakthrough for her came in a second sabbatical at Oxford (1970-71), where she learned a new technique called “chromosome banding”. Scientists there had learned how to differentiate segments of chromosomes by staining them with various dyes, and the pictures of chromosomes now became far more insightful. The banding technique provided a road map for each arm of each chromosome. It was like having signs along a country road, telling you which road you were on and where you were on it.
Rowley turned her attention to studying the chromosomes of cancer cells. Still working part-time, she would bring home micrograph pictures of the stained chromosomes, cut out each chromosome and line them up, sitting at her dining room table. Her children would sometimes tease her about getting paid to play with paper dolls. She later described her studies of chromosomes as a “fishing expedition”, something that scientists are often a little sensitive about admitting to. But she had become an expert angler, and had a sense of where the fish were.
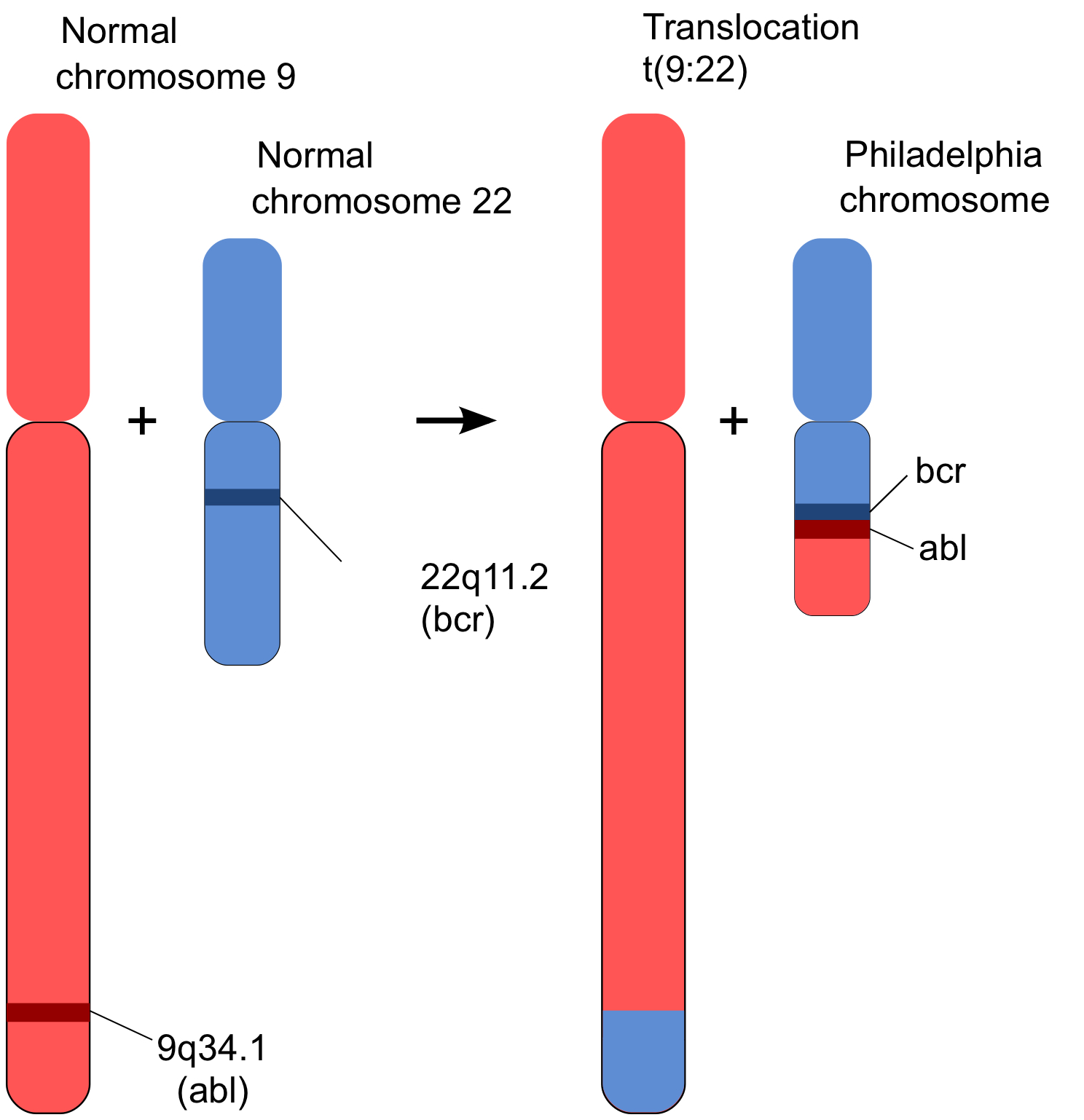
On a fateful day in 1972, Rowley recalled years later, she “lined up the chromosomes from leukemia cells on a table and told my kids not to sneeze”. She spotted several chromosomal abnormalities, and in particular, she suddenly realized that the understanding about the “Philadelphia chromosome” of CML was not right, or at least, it was incomplete. The Philadelphia chromosome wasn’t shorter because it had simply lost part of one of its arms; it had actually exchanged part of that arm with another chromosome, number 9, and gotten in return an even shorter chunk of one of 9’s arms (1). Chromosome 9 now carried a bit of the missing part of chromosome 22. Since 9 was much larger than 22, that addition hadn’t been noticed. But little chromosome 22 had become noticeably shorter by the unequal exchange.
Figure 2 represents what had happened graphically (from here) . Geneticists refer to such an event as a “translocation”. A short bit of chromosome 9 had translocated to chromosome 22, and in return, a part of 22 had been added to 9. An exchange of arms. What turned out to be critical was that the translocated gene from chromosome 9, called “abl” (pronounced “Able”; more about that gene below) was profoundly important in cellular function. And, that the place where it joined chromosome 22, linked to a gene called “bcr” (which stands for “breakpoint cluster region”), had a big effect on those functions. Exactly why the translocation, called BCR-ABL, happens the way it does isn’t understood. But the result of that event is, as will be described in the following section.

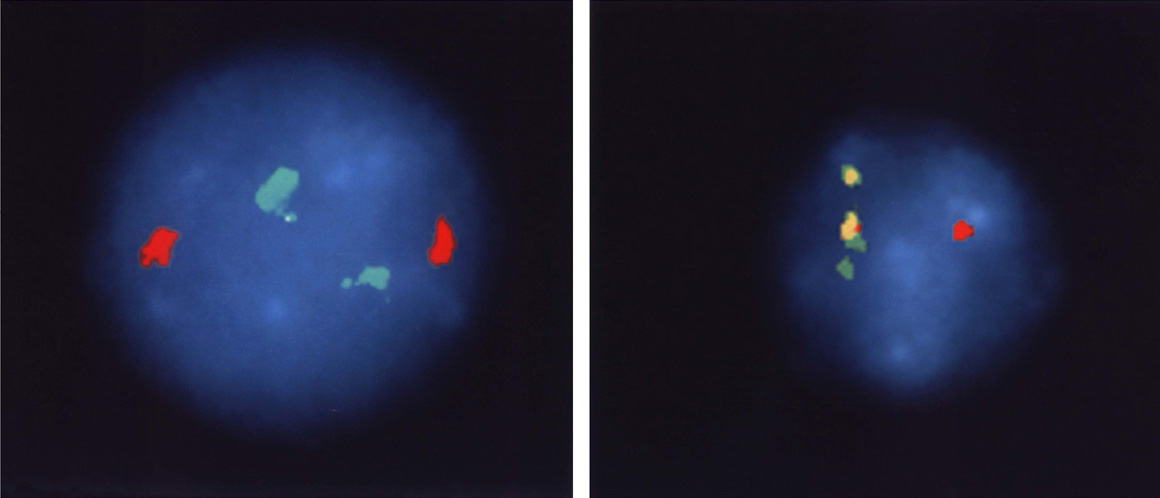
The correctness of our understanding of the translocation event is further supported by the images in Figure 4. Chromosomal sequences bcr and abl (the green and red markers in Figure 4, left panel) are well separated in normal cells because they are on different chromosomes. In CML cells, chromosome 22 carries both genetic segments, and they are so close together the stain is now yellow (the mix of red and green stains located side by side).
What the translocation 9:22 means
Timing is as significant in research as it is in show business. While Janet Rowley was uncovering the novel cytogenetic structure of the translocation between chromosomes 9 and 22 in CML, there was a noisy revolution going on in cancer research. The Philadelphia chromosome story would soon benefit from that revolution. In the 1970s, the favoured theory about how cancer was caused was that viruses were responsible. A number of viruses that caused cancer in animals had been identified, beginning in 1904 with the Rous Sarcoma Virus, RSV. This virus, named after its discoverer Peyton Rous, caused a sarcoma in chickens. A couple of other animal cancer viruses were subsequently identified, and in the 1970s human cancer viruses were expected to soon step into the spotlight. It would surely be only a matter of time before viruses responsible for many human cancers were identified.
Then there was a pause, and finally a complete halt to the idea that human cancer was generally caused by viruses (some are, but most aren’t). Ingenious and painstaking research had shown that RSV contained a cancer-causing gene all right, but that gene was present, in nearly identical form, in all cells of a normal chicken. Long story short, the RSV cancer gene was a mutated form of a normal gene. This pattern was then found in other oncogenic (cancer-causing) viruses, including one that caused a mouse sarcoma, called the Abelson virus after its discoverer, John Abelson. The cancer gene in that case was named “abl” (“able”). Again, a very similar version of abl was found in the genomes of normal mice. The story that emerged was that the cancer-causing viruses had, probably by singular chance, captured a normal gene when they infected animals. This gene had then undergone a mutation that made it an oncogene—a gene that caused cancer. Such viruses were found to be particularly prone to mutations, for reasons that we now understand very well (it’s at the heart of the hypermutability of HIV). When the virus containing the oncogene infected a healthy animal, cancer resulted. The viruses were secondary; the main story was, that cancer could be caused by mutated normal genes. This is the story that cancer biologists worked out, and they are sticking to it.
The insight into the nature of CML was revealed when scientists used their new cancer-gene probes to look at the translocated chromosome 22. The unexpected observation made by a student in a research laboratory in California in 1983 was that the junction between the bit of chromosome 9 that was translocated onto the amputated chromosome 22 in CML (Figure 2) contained the abl gene, which is known to cause cancer in mice in its mutated form. Was it doing the same in CML patients? And what exactly was the abl gene doing, in normal cells and when it led to cancer?
The nature of abl
As part of the revolution in cancer research of the mid-1970s, it was discovered that abl, and other cancer genes, encoded a new type of protein. It was a relative of a known family of proteins, with a twist. All the members of this lineage are enzymes, biological catalysts, that transfer phosphate groups from adenosine triphosphate (ATP) onto proteins. This is the “kinase” family of enzymes. The kinases known at the time abl and src were being described all targeted the amino acids Serine and Threonine. That is, they transferred a phosphate group from ATP onto the hydroxyl groups (—OH) of serine and threonine in specific proteins. What was new about the oncogenes src and abl was that they encoded kinases that transferred a phosphate group onto the amino acid Tyrosine, a rarer amino acid than Serine and Threonine. The phosphate group activates the recipient protein to do whatever it is designed to do in the cell. In the case of the Abl protein (by convention, genes are written in italics, their protein products in normal font), that function includes causing the cell to grow and divide into two cells. The Abl protein is a switch that starts cell proliferation. Normally, Abl does its job and then quiets down as its phosphate group is automatically removed by another part of the regulatory circuit. But the mutated Abl found in the CML genome doesn’t quiet down; it remains fiercely active and continues to drive cell growth. That uncontrolled cellular proliferation is the very heart of cancer.
The causative mechanism of CML was now understood at the molecular level: a translocation between two chromosomes caused a gene for a tyrosine kinase, abl, to relocate to a part of the genome where it was constitutively expressed and active. And this kinase caused the uncontrolled growth of the cell, eventually leading to a tumor.
The path to this understanding benefitted from various lines of scientific discovery: it began with a simple baffling observation (Hungerford and Nowell), it continued with what Janet Rowley herself called “a fishing expedition” that uncovered the true nature of that original observation; and it was rapidly accelerated by a river of new knowledge from molecular research that identified cancer as a disease of the genes.
Describing the mechanism of CML at the molecular level is a wonderful achievement, one of the best examples of the power of molecular biology to help us understand cancer. The next important question was: can this understanding be useful, can it lead to a cure, or at least a treatment for CML? The results are implied by the title of the next post: “Chronic Myeloid Leukemia: A Molecular Solution“.
Go To Contents
Sources
- Rowley, J. 1973. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 243:290 – 293.
- Reid, A., S. M. Gribble, B. J. Huntly, K. M. Andrews, L. Campbell, C. D. Grace, M. E. Wood, et al. 2001. Variant Philadelphia translocations in chronic myeloid leukaemia can mimic typical blast crisis chromosome abnormalities or classic t(9;22): a report of two cases. Brit. J. Haematol. 113:439-442.


{kind=link}