

Dr. Brian Druker and Ms. LaDonna Lopossa, one of the earliest patients to benefit from Gleevec
Rob Schick is the Branch Manger of the Baird Wealth Management Group in Portland, Oregon. Most people would probably consider him lucky. For starters, Portland is one of the most desirable cities in the United States to live in. He and his wife have three grown children, one of whom works at the same company. In addition to his success in business, Rob Schick’s efforts on behalf of the Knight Cancer Institute, where he has helped raise millions of dollars for research, were nationally recognized in 2015 by a National Community Service Award from the Invest in Others Foundation. But dwarfing these considerations, he is lucky to be alive and healthy: in 2000, he was diagnosed with Chronic Myeloid Leukemia (CML), a disease that historically had a 5-year survival rate of less than 50%. But he became one of the early patients enrolled in a new treatment protocol that has had an astonishing success rate. The development of the drug involved, called Gleevec, is based on research which determined the apparent cause of CML—namely, the presence of an overly-active protein kinase that drives the growth of the CML cancer cells. That research is described in the post ” Chronic Myeloid Leukemia: A Molecular Diagnosis“, and also in a very readable book by the science writer Jennifer Wapner (1).
The protein responsible for CML
As described in the previous post, a sequence of basic discoveries led to the conclusion that the growth of cancerous cells in CML was driven by an abnormally active protein kinase. Protein kinase enzymes transfer phosphate groups from the common “high-energy” carrier ATP to side chains of other proteins. This is an important form of cellular regulation, involved in activities ranging from glucose metabolism to gene regulation and cellular proliferation.
An exchange of genetic material between human chromosomes 9 and 22, which is associated with CML, leads to the formation of the protein kinase bcr-Abl. (By convention, the labels of genes are in italics, their protein products in plain text.) The microscopically-visible altered chromosome 22 is called “The Philadelphia Chromosome”, and is diagnostic for CML cells. The rest of the cells in the body have normal chromosomes 9 and 22.
The abl gene is named for John Abelson, the scientist who discovered that it caused cancer in mice; the “bcr’” part of the name stands for “breakpoint cluster region”, the region in human chromosome 22 where the abl gene relocates during the genetic exchange that leads to CML. To summarise (2), the chromosomes of human CML cells typically contain an altered form of the cellular abl gene, bcr-abl. The bcr-Abl protein that this gene codes for (below) is an over-produced and over-active form of the Abl protein tyrosine kinase, and it drives the proliferation of cancer cells in CML.

The conclusion that bcr-Abl kinase is responsible for CML was supported by finding that introducing the bcr-abl gene into mouse cells turned them into cancer cells (3). A smoking gun. The question then arose, could the bcr-Abl protein be a target for chemotherapy of CML? In other words, if the bcr-Abl kinase could be inhibited, would the proliferation of the cancer cells of CML stop? It was a good idea.
The chemists do their thing
While the sequence of causes and effects that linked the Philadelphia chromosome to CML was being worked out, an activity that was initially only tangentially related to it was taking place at the Swiss pharmaceutical company Ciba-Geigy. By the mid-1980s, the importance of protein kinases in cellular regulation was well understood. There are literally hundreds of protein kinases in the cells, each with a unique set of protein targets for phosphorylation, and a specific regulatory role in the cell.
A group of scientists at Ciba-Geigy thought that inhibitors of specific protein kinases might be useful pharmacological agents, and also tools for understanding disease. But their idea was not unchallenged: they sought inhibitors that acted on specific kinases, not on all of them or even a large group. Many scientists, including many of their colleagues, thought that goal was unrealistic. All protein kinases use ATP as the phosphate donor for the kinase reaction, and their enzymatic reactions are similar. It seemed unlikely that one kinase, or even a small subset, could be inhibited uniquely.
A number of biological systems were available to test the usefulness of potential kinase inhibitors. These systems ranged in complexity from purified enzymes to more complicated cellular or sub-cellular systems that carry out various biological processes. They were developed during a productive period, beginning in the early twentieth century, of studying complex biological systems “in vitro“, literally, in glass (although admittedly many of these studies are carried out “in plasticus“). For example, parts of the immune response could be reproduced in cell culture in the laboratory, and its components teased apart. The immunosuppressive agent cyclosporine was discovered at the Swiss pharmaceutical company Sandoz using such systems, and that discovery opened up the field of organ transplantation. The development of in vitro systems was as important for experimental medicine as the development of molecular genetics.
Where to begin looking for potential kinase inhibitors? There are natural products that specifically inhibit some kinases, which provided clues. The natural product inhibitors worked by competing with ATP in binding to the target kinase, and the ATP “binding pocket” on the kinase was thought to be a good target for synthetic inhibitors. A wide assortment of moderately-sized organic molecules with some resemblance to ATP were screened for inhibitory activity. When one was found, it was chemically modified and the effects of modification on its specificity and inhibitory power were studied in vitro (4). It wasn’t a subtle approach, more of a brute-force effort, but it could be powerful if pursued with enough resources, which the pharmaceutical lab had.
The chemists reach out
By 1986 the list of potential kinase inhibitors was growing, but the Ciba-Geigy scientists needed help with more sophisticated assays for their biological activities. A few members of the team, including Dr. Nick Lydon, travelled to the Dana Farber Cancer Institute in Boston to ask scientists there for help setting up more sophisticated bioassays for their inhibitors. The Ciba-Geigy scientists returned to Switzerland with new research tools, but also an important personal connection. One of the young MD-scientists at the Dana Farber Institute, Brian Druker, was excited about the possibility of using kinase inhibitors to attack cancer cells, in particular, CML. The bcr-Abl kinase, which appeared to be the driving force for the proliferation of CML cells, might be a target for such an inhibitor. However, discussions about further collaboration between the Dana Farber Institute and the Ciba-Geigy team were moot because the Farber Institute had recently formed an exclusive agreement with Sandoz, Ciba-Geigy’s rival across the Rhine river in Basel.
Beginning with an organic chemical that inhibited one class of kinase (not the class to which the Abl kinase belonged), the chemists added various side chains to it, hoping to increase it potency. To their surprise, one of these chemical modifications resulted in the inhibitor switching its affinity; now it did inhibit the Abl kinase, although it no longer worked on the kinase it originally blocked. This new compound was made water soluble, so that it could be taken orally if it panned out. It was identified as STI571 in the arcane vocabulary of the chemistry lab, This was exciting news for Dr. Druker, because he had been urging the Ciba-Geigy chemists to look for inhibitors of Abl kinases. By 1993, he had moved to the Oregon Health and Science University in Portland, Oregon, and was ready to pursue the exciting possibility of using kinase inhibitors to stop the growth of CML cells.
In addition to the bcr-Abl version of the kinase that is found in CML cells, there is also a normal cellular version of it which lacks the bcr part. It has a role in normal cellular physiology. There is also a form that is produced by the Abelson Leukemia Virus of mice, which had led to the original identification of abl as a cancer-causing gene, or oncogene. STI571 inhibited all three forms of the Abl kinase. What would it do to the proliferation of CML cells? And, would it be too toxic for human use because it also inhibits normal Abl kinase, at least partially?
Druker tested STI571 on cells that had been transformed by the bcr-abl oncogene in the laboratory. These are otherwise normal cells that have had the bcr-abl gene introduced by genetic engineering. As hoped, STI571 inhibited their proliferation. Even more exciting, it also prevented the growth of tumor cells expressing the oncogene in mice, and when bone marrow cells from patients with CML were cultured in vitro, the tumor cells were strongly inhibited from proliferating. STI571 specifically inhibited growth driven by bcr-Abl; cells that grew in response to growth stimulating hormones, or because they were expressing a different oncogene, were unaffected, as were normal cells. Druker and Lydon and their collaborators published these first, exciting results in 1996 (5, 6). As it became clear that STI571 was at least worth studying as a possible drug, it was given the more mellifluous name “Gleevec” (in Europe it was called “Glivec”). Its generic name was imatinib.
The structure of Gleevec was determined by X-ray crystallography. It bears a passing resemblance to ATP. Gleevec, and other inhibitors of bcr-Abl kinase that were discovered subsequently, works by binding competitively to the same part of the Abl kinase that ATP normally occupies. It is a competitive, efficient, inhibitor of ATP binding.
The structure of the complex formed between Gleevec and the Abl kinase has also been determined by X-ray crystallography, and it confirms that the drug binds in the same part of the protein that is occupied by ATP during its kinase action (7). Gleevec binds an inactive conformation of the bcr-Abl kinase and thereby prevents its auto-phosphorylation (the kinase first adds a phosphate group to itself to become activated).
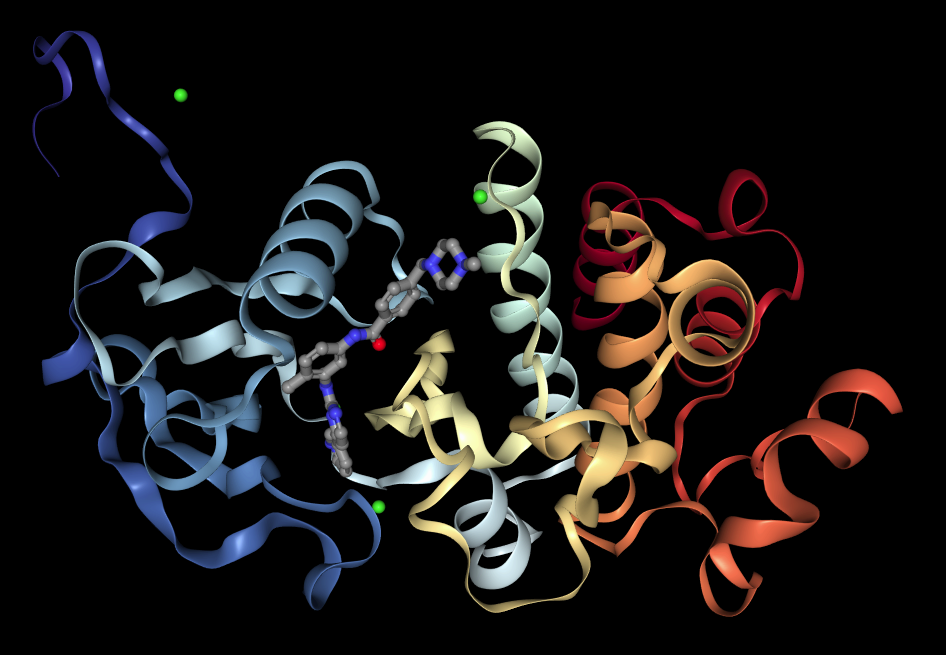
An even more revealing, three-dimensional representation can be found at this web site. You can get a good impression of the 3-dimensional structure of the molecule by clicking on the button labelled “Spin”. Notice how intimately Gleevec contacts parts of the protein. Binding is tight, inhibition is strong.
The pharmaceutical business is a business
In the normal course of drug development, such a promising initial laboratory result might be followed by years of further testing in vitro, and even more years of clinical trials through Phases One to Three. But, after a bit of a hiccup, Gleevec experienced a much faster gestation.
Progress toward its development initially bogged down, in part because Novartis was reluctant to put up the money needed for clinical testing of Gleevec. This may seem counter-intuitive, given the dramatic results from the lab. But the costs of clinical development are high, typically exceeding the costs of the original research by more than ten-fold. Taking a new drug through three phases of clinical trials over many years would probably cost hundreds of millions of dollars, and the risk of failure was high, as it is with any new candidate drug. There were also early signs of toxicity in dogs and rats. And even if it all went well, the market appeared to be small; only about 6,000 new cases of CML a year are diagnosed in the USA, for example. Furthermore, Novartis at that point had gotten away from trying to find new cancer drugs. Its main interest was related to the development of powerful immunosuppressive drug such as cyclosporine, which had been discovered at Sandoz. And as already described, many of the Novartis scientists and executives were skeptical about the ability of a kinase inhibitor to work without devastating side effects caused by the inhibition of other kinases, including the normal Abl kinase. The view of a company insider on these problems can be found in the book “Magic Cancer Bullet. How a Tiny Orange Pill is Rewriting Medical History” (8).
Druker begged Novartis to support clinical testing of Gleevec, even offering to take the compound off their hands to find other funding for its development. Eventually the company agreed to support the work. The payoff came with breathtaking speed; there were immediate beneficial effects for CML patients. While Phase One clinical tests are designed to test for safety, not efficacy, some of the first patients enrolled in the Gleevec Phase One test in 1998 clearly were benefitting. Partly because CML had such a dire prognosis and there was no effective therapy, FDA approval for Gleevec was received in 2001, just 5 years after the first published in vitro results with CML cells.
Furthermore, the market for Gleevec was much larger than initially thought. In contrast to conventional chemotherapy drugs that kill cancer cells and are used only for a brief treatment period, Gleevec needs to be used continuously to keep CML in remission. The first word of the cancer’s name is ‘Chronic’, but until there was Gleevec, CML was chronic in name only. Without effective treatment it moves to an accelerated phase and blast crisis, where the proportion of cancer cells increases dramatically, and the cancer becomes malignant. Gleevec controls the disease, but it must be taken continuously, probably for the life of the patient (it’s too soon to know for certain). In other words, with Gleevec, CML became a chronic disease in fact as well as nominally. The pharmaceutical industry had created an ever-increasing market, in the form of a surviving cohort, for one of its products where initially it appeared to be very small (9).
The story does not end there
Although the story to here has been little short of miraculous, it isn’t clear sailing. Several important issues remain. In the first place, many patients develop resistance to Gleevec. Gleevec resistance is often caused by a single genetic mutation in the abl part of the bcr-abl gene. Figure 1 shows the amino acid sequence of one bcr-Abl kinase, and the difference in one Gleevec-resistant clone of CML cells (the “E” (glutamic acid) residue shown in red in row 8).
After the FDA approval of Gleevec, pharmaceutical labs identified “second-line” treatments for CML that acted similarly to Gleevec. This group includes Dasatinib (Sprycel) and others (there are now 5 drugs either approved or in development). Dasatinib is used when the bcr-abl gene has mutated to produce a Gleevec-resistant kinase. It is very potent, and can be used for patients who do not tolerate Gleevec. Constant monitoring is needed to watch for resistance.
The 5-year survival rate after early use of Gleevec is 88%, but as CML progresses from chronic to accelerated and blastic phases there are additional mutations in other oncogenes, which may explain why Gleevec doesn’t work as well at these stages. At the latter stages of the disease, the Abl kinase may not be essential, and Gleevec will therefore not be effective.
Another issue is that the drugs need to be taken continuously. The stem cells in the bone marrow that give rise to the CML cells in the blood are not wiped out by the drugs. Or at least, not immediately or usually. Monitoring of survivors may determine if they are ‘cured’ at some point, but until then, they need to continue to take the drugs, which are very expensive. Novartis, which originally thought STI571 was unlikely to pay off, now earns, worldwide, over 4 billion dollars a year from its sales.
Gleevec is a paradigm for future approaches to cancer treatment. Dozens of other cancers are being studied to see if a similar ‘magic bullet’ can be found. A difficulty is that while CML is a relatively simple disease, driven by one dysfunctional gene, most cancers involve several mutations, and may therefore be more difficult to treat. But at least we have an example of how things can turn out if we can find the key.
Gleevec has also proved to be useful in treating people with gastrointestinal stromal tumors, GIST. These tumors, often growing in the lining of the stomach or intestines, are usually fairly large when they are discovered, and must be removed surgically. Conventional chemotherapy is ineffective against GIST, but Gleevec, it turns out, prevents regrowth both at the primary site after surgical removal and at sites in other parts of the body (metastases).
GIST is most often caused by a mutation in one of two cellular signalling molecules. One of these is called PDGFR, the other c-kit. Both of them have an intracellular protein kinase as part of their structure, linked to a receptor component outside. The c-kit kinase is a homolog of a viral oncogene first discovered in cats! Gleevec, in addition to inhibiting the activity of the bcr-Abl kinase of CML cells, also inhibits the over-active c-kit kinase and the PDGFR kinase of GIST. Gleevec also inhibits certain other, non-cancerous, hematological disorders caused by other receptor-kinases. Although longer-term survival data are not yet available, the results point to useful effects of Gleevec here, too.
The survivors’ stories
At the beginning, I described the remarkable change of fate for Rob Schick, one of the early beneficiaries of Gleevec at the Oregon Health Sciences University. Schick is a patient of Dr. Brian Druker. Dr. Druker described an even earlier participant in the clinical trials in 2007 (2). The patient was a 55-year-old man diagnosed with CML. He was treated with the available methods, but over time his clinical findings showed that he was losing to his cancer. In 2000, he was enrolled in a clinical trial of Gleevec. His cancer regressed, immediately and dramatically. But over time, it began to grow again. The bcr-abl gene had mutated and become Gleevec-resistant. He was quickly enrolled in a trial for the “second-line” drug Dasatinib. Dr. Druker described what happened:
“Over the last 3 years, he [the patient] has been treated with Dasatinib and has maintained a major cytogenetic response [i.e. CML cells have disappeared from his blood]. In February of 2007, this male reached a full decade of living with CML, during which he has been able to continue to work and maintain his normal lifestyle, whereas previously, survival beyond five years of patients with CML was uncommon.” (2).
Another survivor is LaDonna Lopossa, shown at the top of this post. She was suffering profoundly from CML, and had resigned herself to dying when she was enrolled in the first clinical trial of Gleevec in early 2000. She has said that she knew, within a week, that Dr. Druker was onto something transformative, as she began to regain her health. She is alive and well today, despite her CML having developed resistance to Gleevec. She is on her fourth drug to keep it in remission.
Go to Latest Posts
Sources
- Wapner, J. 2013. The Philadelphia Chromosome. The Experiment, LLC, New York, NY.
- Sherbenou, D. W., and B. J. Druker. 2007. Applying the discovery of the Philadelphia chromosome. Journal of Clinical Investigation 117:2067-2074.
- Kelliher, M. A., J. McLaughlin, O. N. Witte, and N. Rosenberg. 1990. Induction of a chronic myelogenous leukemia-like syndrome in mice with v-abl and bcr/ABL. Proc Natl Acad Sci U S A 87:6649-6653.
- Traxler, P., G. Bold, E. Buchdunger, G. Caravatti, P. Furet, P. Manley, T. O’Reilly, et al. 2001. Tyrosine kinase inhibitors: from rational design to clinical trials. Medicinal research reviews 21:499-512.
- Buchdunger, E., J. Zimmermann, H. Mett, T. Meyer, M. Muller, B. J. Druker, and N. B. Lydon. 1996. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res 56:100-104.
- Druker, B., S. Tamura, E. Buchdunger, S. Ohno, G. Segal, S. Fanning, J. Zimmermann, et al. 1996. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of bcr-Abl positive cells. Nat Med 2:561 – 566.
- Nagar, B., W. G. Bornmann, P. Pellicena, T. Schindler, D. R. Veach, W. T. Miller, B. Clarkson, et al. 2002. Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res 62:4236-4243.
- Vasella, D., and R. Slater. 2003. Magic Cancer Bullet. How a Tiny Orange Pill is Rewriting Medical History. HarperBusiness/Harper Collins, New York.
- Mukherjee, S. 2010. The Emperor of All Maladies. A biography of Cancer. Scribner, New York.



{kind=link}