
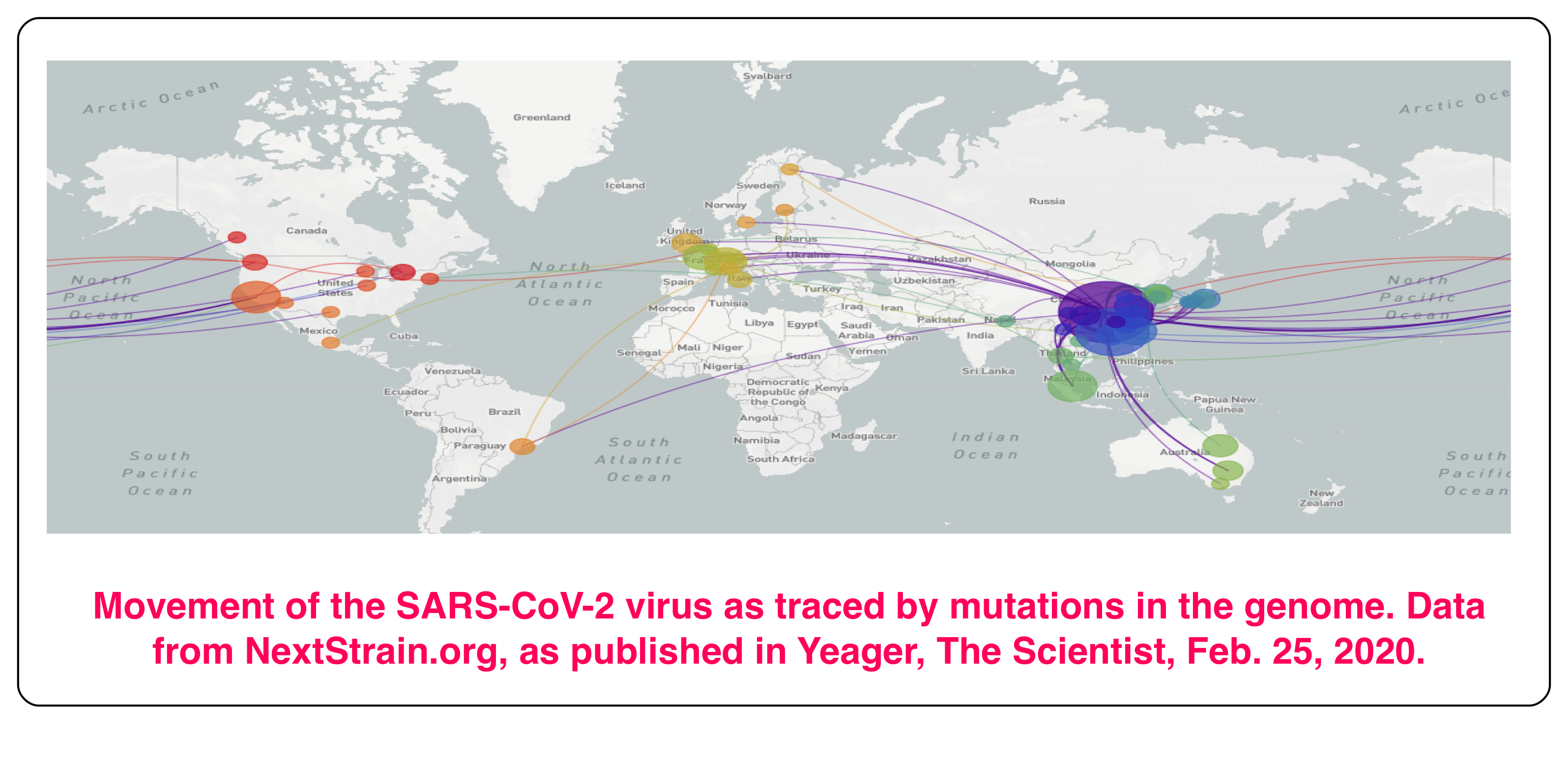
As of March 4, 2020 the novel coronavirus SARS-CoV-2, which causes COVID-19, has spread to more than 77 countries. More than 93,000 cases have been reported, and just under 3,200 deaths. It’s thought that the virus infected its first human victim in the city of Wuhan in China, in November, 2019. That case was apparently caused by transmission of SARS-CoV-2 from an animal. Since then, it has spread by human-to-human contact. The virus probably originated in bats, and then likely passed through another animal before it found its human host. Along the way from bats it underwent changes, mutations, that made it transmissible to, and by, humans.
Much of what we know about the global spread of SARS-CoV-2 comes from genetic analysis of isolates from around the world. All of them appear to be derived from a single infection in Wuhan. There are differences in viral genomes taken at different times and places, and these provide useful information about the relationships between different outbreaks. But genetic analysis also identifies the critical event that allowed the virus to jump from its presumed reservoir, the bat, to an animal, and then to humans.
Update: According to an article in the New York Times on April 9,”New research indicates that the coronavirus began to circulate in the New York area by mid-February, weeks before the first confirmed case, and that travelers brought in the virus mainly from Europe, not Asia.” This was the conclusion of several labs studying the genomes of the virus.
The basis for the steady accumulation of mutations in SARS-CoV-2 lies in the mechanics of its replication. The principles involved are also applicable to other RNA viruses, such as the Human Immunodeficiency Virus, the earlier coronavirus infection caused by SARS, and influenza. The drifting genetic makeup of these viruses is a factor in attempts to block viral infection.
DNA Genomes Are Stable
The DNA of animal genomes is heavily protected against mutational change. Firstly, it is double-stranded, as proposed in 1953 by Watson and Crick. Those two burst into their favourite Cambridge pub at noon on a Saturday in February of that year to announce to the world that their double helical DNA model revealed “the secret of life” (in Watson’s recollection, it was Crick who said that; Crick claims not to remember doing it). While that would certainly have been an overstatement, their model for DNA was an incredible advance in understanding how genes worked. For one thing, the two strands are strictly complementary. Because of that, a change in a base in one strand is immediately detected as damage, and corrected.
Changes in DNA, genetic damage, can happen while it’s being copied (“replicated”) in preparation for cell division, or during other parts of the cell cycle. A number of agents can cause genetic damage: external agents such as X-rays or genotoxic chemicals such as nitrates, or agents generated inside cells, such as oxygen free radicals. Almost all changes in DNA are quickly repaired, by a variety of mechanisms. Only a tiny fraction escape repair, and become mutations. It is estimated that a mammalian cell undergoes some one million DNA-damaging events a day, but only the rare one becomes a permanent change in the cell’s DNA, a mutation. Maintaining the integrity of the genome is very important, and at least 168 genes have been identified that are involved in the correction of DNA damage. RNA genomes, on the other hand, lack most of the error-correcting mechanisms that DNA is protected by, and they consequently mutate more rapidly.
RNA Viruses Are More Susceptible to Mutation
When cells divide, or when viruses proliferate inside living cells, an existing template, either DNA or RNA, is copied by one or another of the polymerase enzymes. This copying happens with very few mistakes. The template guides the synthesis of new strands by the Watson-Crick rules of pairing. There’s a slight difference between RNA and DNA in these pairing rules: DNA uses the base pairings A with T, and G with C. RNA has two chemical differences from DNA. Its sugar is ribose, whereas DNA has the sugar deoxyribose. And RNA doesn’t contain T (thymine); is uses the base U (uracil), which has the same pairing properties as T (it pairs with A), but doesn’t have the methyl group on its pyrimidine ring that thymine has.
Although the coding properties of T (in DNA) and U (in RNA) are identical, the presence of U in RNA is one of the reasons for its relative instability, compared to DNA. A common genome-damaging event is deamination of the base cytosine (C). That is, the removal of an amino group, which can be caused by chemical or physical genotoxic attack. With the removal of the amino group, C becomes U. C and U differ only by that amino group. DNA does not contain U, and there are enzymes and proteins in the cell that stand guard for its occurrence. These enzymatic mechanisms detect U (uracil) as soon as it is formed by the deamination of C, and quickly replace it with a C residue. Damage repaired. But RNA normally does contain U, so the generation of another U by deamination of C doesn’t register as a damaging event; naturally, there is no mechanism to detect and remove U from RNA, since it is a normal component. Because of that, events that generate U from C in RNA will remain unrepaired, that is, they produce a C-to-U mutation in the RNA molecule.
There are many roles for RNA in normal cell biology — for example, that of messenger RNA, mRNA, which carries genetic information from the gene to the protein synthesizing machinery. But there are hundreds, or even thousands, of copies of each evanescent mRNA in the cell. So the occasional conversion of C to U in mRNA doesn’t matter. RNA performs a variety of roles, and a chemical change in one or a few molecules of cellular RNA is not significant. But an RNA virus has a single RNA molecule as its genome, and a change in that RNA molecule produces a mutated virus.
There are other reasons for the instability of RNA genomes. For poorly understood reasons, single stranded nucleic acids, such as the RNA genome of SARS-CoV-2, are more mutable than double-stranded DNA (or RNA) molecules. More importantly, the enzymes that synthesize a new strand of RNA, RNA-dependent RNA polymerases (there are several kinds, but all share this property), don’t have the copy-editing function of the DNA-dependent DNA polymerase that replicates the DNA genome. When a DNA polymerase mis-incorporates a base during DNA synthesis, which can happen to any polymerase, that newly incorporated base doesn’t fit comfortably into the double helix. The DNA polymerase senses this malformation, and it backspaces to repair the error. But RNA polymerase goes blithely on after a mis-incorporation, with the incorrect base now part of the new strand of RNA. This polymerase doesn’t have spellcheck, and the misincorporated base stays. Mutation.
Almost none of the 168 or so gene products that guard the DNA genome from damage have a parallel in the RNA world. RNA looks like a more primitive genome. (Many scientists believe that the first replicating genomes were RNA, and that the RNA world eventually gave rise to the DNA world.) The RNA viruses are more mutable, and less protected, than DNA in cells, or DNA viruses. This is part of the reason that the genomes of RNA viruses are usually small, not much more then 10,000 bases in length. Many mutations are deleterious, and it’s thought that large RNA genomes in viruses would become unstable because of their high mutation rate. So most RNA viruses have a fairly short genome. HIV’s RNA genome is about 9,200 bases long, whereas there are DNA viruses with more than 300,000 bases.
The genus to which SARS-CoV-2, influenza, and the common cold belong is something of an exception. Its members do have some genes that code for error correction, and it’s thought that their relatively larger genomes are a result of this. The coronavirus genome codes for at least one protein that carries out the “spellcheck” function missing in the RNA polymerase, and its mutation rate per base is lower than it is for simpler, smaller viruses. The SARS-CoV-2 genome is almost 30,000 bases long, a length that is thought would result in too many errors to be viable without the repair functions it encodes. But even so, it still mutates at least 100 times faster than the cells it infects.
The Goldilocks Principle Strikes Again
There’s no doubt that mutability can be advantageous to RNA viruses. The conversion of SARS-CoV-2 to an infectious agent for humans resulted from mutation. The cellular target for SARS-CoV-2, the molecule it attaches to on the outside of the cell to start an infection, is known: it is Angiotensin Converting Enzyme (ACE2). This is the case for humans and other animals that SARS-CoV-2 can infect. But in bats, the presumptive SARS-CoV-2 precursor (greater than 90% identical to SARS-CoV-2) has a different cellular target. It was necessary for the viral attachment protein to mutate to become infectious in animals other than bats.
When SARS emerged in 2003, it quickly adapted to bind human cells with greater affinity/infectivity. It seems likely that viruses mutate as quickly as is consistent with successful spread in the target population. If they mutated much quicker, the generation of damaging mutations would limit growth and could lead to extinction. On the other hand, too slow a rate of mutation limits the spread of the virus.
So What?
So, there it is. RNA viruses, including SARS-CoV-2, mutate relatively quickly, and this makes them at once more dangerous, and also easier to track. Eight mutations have been found in the 30,000 bases of the 161 fully sequenced SARS-CoV-2 genomes by the beginning of March 2020. The different mutant forms can all be traced back to the original infection in Wuhan, and the points at which new versions occurred can be identified. It’s very similar to what biologists do when they build an evolutionary tree, with branches wherever a new path of evolutionary development starts. This has provided a trace for the infections seen in different parts of the world, as shown in the figure at the top of this post.
It was this kind of analysis which uncovered the fact, made public at the beginning of March, that two people in Washington state had undoubtedly been infected by the same strain of SARS-CoV-2. Their cases were weeks apart, and they had not come in contact with each other. Both carried a mutation that had been seen only twice in the Chinese samplings, so it was unlikely to have arisen twice, independently, in Washington. The disturbing implication was that the virus was circulating, undetected, in other people, who remained asymptomatic. To uncover such underlying infection, the US FDA has given permission to institute much more widespread screening for characteristic genetic markers of SARS-CoV-2. The screening depends on the DNA profiling technique using the Polymerase Chain Reaction described in earlier posts (here and here).
One result of the mutability of viral RNA genomes is that they can become resistant to drugs or antibodies. For example, each winter we are advised to become immunized against the current version of ‘flu. But the current version isn’t known precisely; it is partially a guess, and depends on what has been seen recently. It is usually quite different from last year’s version, because its coat protein, the target of immunisation, is different: the gene coding for it has been selected from a pool of variant forms by Darwinian selection.
Similarly, drugs that attack the reverse transcriptase enzyme of HIV can eventually lose their potency, when the DNA sequence encoding the enzyme changes slightly in a way that doesn’t knock out its functionality, but does make it resistant to the drug. Mutations are often deleterious, of course, and disappear by natural selection. But when a mutation provides an advantage in the current environment, such as the presence of a drug that attacks the virus, it can take over. The strong survive.
The Future
Currently, the best estimates are that a vaccine against SARS-CoV-2 will take a year or more to develop and test, if it is even possible. Development of an antibody vaccine against HIV has not yet succeeded after decades of work. And there’s always a chance that an antibody that is effective at one point will lose its power due to mutation of the target protein (as in seasonal ‘flu).
There is at least one drug (remdesivir) currently being screened for effectiveness against SARS-CoV-2. It was partially developed in response to the SARS epidemic of 2003, but never brought to completion because the virus disappeared as a result of conventional isolation techniques. The hope is, that drugs stopping one coronavirus may halt another, due to the commonality of their biologies. Quick development should be possible because it has already been tested for safety. If clinical trials are successful, remdesivir could be available within months. But as in the case of the restless genome of HIV, a drug that works on a viral target at one point may lose its effectiveness due to genetic change.
Nobody knows what the outcome of the COVID-19 saga will be. Perhaps conventional isolation techniques will work to stop the epidemic, as it did with SARS. But it’s unlikely that vaccines or drugs will help much in the near future, and even after that, genetic instability may make the problem difficult to contain. Blame it on the instability of the RNA genome.
Go To Contents
Sources of Information on SARS-CoV-2
Brennan J. RNA Mutation Vs. DNA Mutation. Sciencing https://sciencing.com/rna-mutation-vs-dna-mutation-3260.html
Duffy, S. Why are RNA virus mutation rates so damn high? PLOS Biology, August 13, 2018. http://doi.org/10.1371/journal.pbio.3000003
Dennison, M. R., Graham, R. L., Donaldson, E. F., Eckerle, L. D., Baric, R. C. Coronaviruses: an RNA proofreading machine regulates replication fidelity and diversity. RNA Biology 8(2):270 (2011).
Yeager, A. Coronavirus’s Genetics Reveal Its Global Travels. The Scientist, February 25, 2020. https://www.the-scientist.com/news-opinion/coronaviruss-genetics-reveal-its-global-travels-67183
Coronavirus disease (COVID-19) situation reports. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports
Boseley, S., Devlin, H., and Belam, M. What is coronavirus and what should I do if I have symptoms? The Guardian, March 4, 2020.
Woodward, A. The genetic code of the Wuhan coronavirus shows it’s 80% similar to SARS. New research suggests a potential way to neutralize the virus. Business Insider, February 3, 2020.
Peck, K. M., and Lauring, A. D. Complexities of Viral Mutation Rates. Journal of Virology, 92:e01031-17 (2018).
Fink, S., and Baker, M. Coronavirus May Have Spread in U.S. for Weeks, Gene Sequencing Suggests. New York Times, March 1, 2020.


